






Medizintechnik Wissensportal
Der Content Hub der TÜV Rheinland Akademie
Im Zeitalter der Digitalisierung sind wir mit einer ständigen Weiterentwicklung von Wissenschaft und Technik konfrontiert und müssen uns ständig neuen Wissensanforderungen stellen. Die Globalisierung der Unternehmen erfordert die weltweite Vergleichbarkeit von Kompetenzen und qualitätsgesicherten Zertifikaten. Ein TÜV-Abschluss in Medizintechnik ist ein solches Zertifikat, das Ihnen einen Wettbewerbsvorteil verschafft und Ihre Karrierechancen erhöht.
Auf dieser Seite erfahren Sie mehr über die Vorteile einer Medizintechnik-Ausbildung mit TÜV-Abschluss und warum ein solches Zertifikat für Unternehmen und Fachkräfte gleichermaßen wichtig ist.
Wenn Sie eine Karriere in der Medizintechnikbranche anstreben, ist ein TÜV-Abschluss ein wichtiger Nachweis für Ihre fachliche Kompetenz und Qualifikation. TÜV Rheinland bietet Ihnen verschiedene Lehrgänge für unterschiedliche Fachbereiche der Medizintechnik an, die Ihnen fundiertes Fachwissen und praktische Fertigkeiten vermitteln. Mit einem PersCert TÜV-Zertifikat für Medizintechnik dokumentieren Sie neutral und unabhängig, dass Sie die hohen Standards und Anforderungen erfüllen, die für die Entwicklung, Herstellung und den Vertrieb von Medizinprodukten gelten.
Wie auch immer Sie sich entscheiden, wir bieten Ihnen in jedem Fall die gleiche Flexibilität bei der Termin- und Ortswahl. Mit unserer umfangreichen Suchfunktion können Sie gezielt Stichworte eingeben oder nach Themengebiet, Ort, Termin und Art der Weiterbildung filtern. Oder nutzen Sie das Tool myAdvice für eine individuelle Seminarempfehlung mit nur wenigen Klicks.
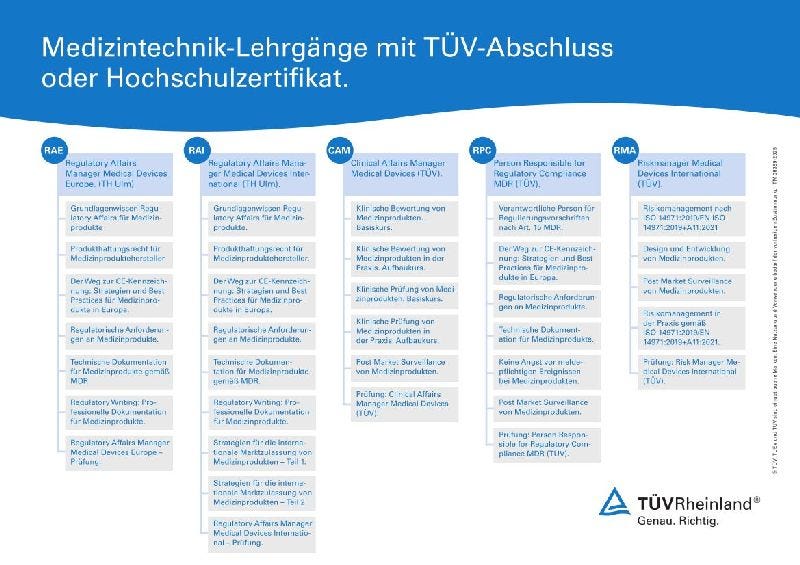
Die enge Verzahnung von Inverkehrbringen und Regulatory Affairs ist entscheidend für den Erfolg in der Medizinprodukteindustrie. Clinical Affairs Manager, Regulatory Affairs Manager, PRRCs und Experten der technischen Dokumentation spielen eine Schlüsselrolle dabei, den komplexen Zulassungsprozess zu steuern und sicherzustellen, dass alle gesetzlichen Anforderungen erfüllt sind. Dies ermöglicht nicht nur einen erfolgreichen Marktzugang, sondern trägt auch zur Sicherheit und Wirksamkeit der Produkte bei, was letztlich das Vertrauen der Nutzer stärkt.
Unsere Lehrgänge und Weiterbildungen im Bereich 'Inverkehrbringen von Medizinprodukten' und 'Regulatory Affairs' informieren Sie über die Anforderungen und Zulassungsverfahren für Medizinprodukte in Europa und weltweit, damit Sie Ihre Produkte sicher und erfolgreich vermarkten können.
Clinical Affairs Manager Medical Devices (TÜV).
Gesamtlehrgang
Die klinische Anwendung von Medizinprodukten erfordert den Nachweis, dass sie in der Praxis den gewünschten klinischen Nutzen erbringen. Dazu werden klinische Daten benötigt, die das Medizinprodukt über den gesamten Lebenszyklus belegen und bestätigen. Clinical Affairs ist für alle Fragen im Zusammenhang mit diesen klinischen Daten zuständig und muss die klinischen Anforderungen der Medical Device Regulation (MDR) erfüllen, die die Sicherheit und klinische Leistungsfähigkeit von Medizinprodukten regelt.
Regulatory Affairs Manager Medical Devices Europe. (Hochschulzertifikat TH Ulm)
Gesamtlehrgang
Der Lehrgang 'Regulatory Affairs Manager Medical Devices Europe.' vermittelt umfassendes Wissen über regulatorische Anforderungen für Medizinprodukte im europäischen Markt (CE-Raum). Die Teilnehmer lernen, den gesamten Markteinführungsprozess zu steuern, die CE-Kennzeichnung zu erlangen und zivil- sowie strafrechtliche Haftungsrisiken zu minimieren. Der Kurs besteht aus sechs Modulen (+ optionale Hochschulprüfung), kombiniert Blended Learning und praxisorientierte Workshops und bereitet auf die Hochschulprüfung "Regulatory Affairs Manager. Europe" vor, die von der Technischen Hochschule Ulm durchgeführt wird.
Regulatory Affairs Manager Medical Devices International. (Hochschulzertifikat TH Ulm)
Gesamtlehrgang
Der Lehrgang "Regulatory Affairs Manager Medical Devices International." vermittelt umfassendes Wissen über regulatorische Anforderungen für Medizinprodukte auf internationalen Märkten. Er deckt alle relevanten Bereiche ab, von der Zulassungsstrategie bis zur Marktüberwachung, und umfasst acht Module (+ optionale Hochschulprüfung). Die Teilnehmer lernen, internationale regulatorische Anforderungen zu identifizieren, Produkthaftungsrisiken zu minimieren und Marktzugangstrategien zu entwickeln. Praxisnahe Workshops und Blended Learning sind Teil des Programms, das auf eine Hochschul-Zertifizierung der Technischen Hochschule Ulm abzielt und besonders für Fachkräfte im Produktmanagement und Regulatory Affairs geeignet ist.
Person Responsible for Regulatory Compliance MDR (TÜV). Gesamtlehrgang oder Kompaktlehrgang
Hersteller von Medizinprodukten müssen gemäß Artikel 15 der EU-Medizinprodukteverordnung (MDR) 2017/745 eine oder mehrere Personen benennen, die für die Einhaltung der regulatorischen Anforderungen verantwortlich sind (PRRC - Person Responsible for Regulatory Compliance). In unserem modularen Lehrgang lernen Sie die Verantwortlichkeiten, Aufgaben und die organisatorische Einbindung einer PRRC kennen. Damit sind Sie in der Lage, diese Rolle zu übernehmen.
Expert Technical Documentation Medical Devices (TÜV).
Gesamtlehrgang oder Kompaktlehrgang
Die Technische Dokumentation ist das wichtigste Dokument für die CE-Kennzeichnung von Medizinprodukten. Sie zeigt, dass das Medizinprodukt den regulatorischen Anforderungen entspricht. Wir erklären Ihnen, wie Sie die Technische Dokumentation klar, strukturiert, vollständig erstellen und über den gesamten Produktlebenszyklus auf dem neuesten Stand halten. Werden Sie zum Experten mit diesem umfassenden Lehrgang der TÜV Rheinland Akademie – auch verfügbar als Kompaktlehrgang!
Unsere Lehrgänge und Weiterbildungen im Bereich 'In-vitro-Diagnostika' zeigen Ihnen, wie Sie die Anforderungen und Neuerungen der IVDR für Ihr IVD erfüllen und umsetzen können, damit Sie Ihr IVD sicher und erfolgreich auf den europäischen Markt bringen können.
In-vitro-Diagnostic Expert (TÜV).
Gesamtlehrgang
Die Entwicklung eines In-vitro-Diagnostikums ist für alle Beteiligten eine komplexe Aufgabe. Die In-vitro-Diagnostics Device Regulation (IVDR) regelt die Zulassung und Überwachung von In-vitro-Diagnostika in der EU und für die CE-Kennzeichnung. In unserem Lehrgang lernen Sie, wie Sie Ihr IVD konform entwickeln, wie Sie die CE-Kennzeichnung und Konformitätsbewertung durchführen müssen und wie Sie Risikomanagement und Gebrauchstauglichkeit über den gesamten Produktlebenszyklus optimal umsetzen.
Person Responsible for Regulatory Compliance IVDR (TÜV).
Gesamtlehrgang
Hersteller von In-vitro-Diagnostika müssen nach der EU-Verordnung über In-vitro-Diagnostika (IVDR) 2017/746, Artikel 15, eine oder mehrere Personen benennen, die für die Einhaltung der Regulierungsvorschriften zuständig sind (PRRC - Person Responsible for Regulatory Compliance). In unserem modularen Lehrgang lernen Sie, welche Verantwortlichkeiten, Aufgaben und organisatorische Einbindung eine PRRC hat. So können Sie diese Rolle übernehmen und Ihren nächsten großen Karriereschritt machen.
Unser Lehrgang und unsere Weiterbildungen im Bereich 'Medizinische Software' vermittelen Ihnen das Know-how, um medizinische Software nach den geltenden Standards zu entwickeln, zu validieren und benutzerfreundlich zu gestalten.
Expert Medical Software (TÜV).
Gesamtlehrgang
Software ist ein wichtiger Bestandteil der Medizintechnik. Die Entwicklung medizinisch genutzter Software muss die gesetzlichen und normativen Anforderungen an Medizinprodukte erfüllen, die Sicherheit und Gebrauchstauglichkeit berücksichtigen und Fehler vermeiden. So muss z.B. die Softwarenorm EN 62304, das Risikomanagement nach ISO/EN ISO 14971:2019/EN ISO 14971:2019+A11:2021 und die Gebrauchstauglichkeit nach EN 62366 eingehalten werden. In unserem Lehrgang erfahren Sie, wie Sie medizinische Software normgerecht entwickeln und dokumentieren können.
Unser Lehrgang und unsere Weiterbildungen im Bereich 'Produktsicherheit und Gebrauchstauglichkeit' vermitteln Ihnen das Wissen, um die Sicherheit und Leistungsfähigkeit Ihrer Medizinprodukte gemäß den aktuellen Normen zu gewährleisten, indem Sie Fehlerpotenziale und Anwenderfehler erkennen und vermeiden.
Medical Devices Usability Expert (TÜV).
Gesamtlehrgang
Medizinprodukte müssen ergonomisch und benutzerfreundlich gestaltet sein, um ihre diagnostischen und therapeutischen Funktionen optimal zu erfüllen. Dadurch können teure Nachbesserungen vermieden und Anwendungsfehler minimiert werden. In unserem Lehrgang lernen Sie, die relevanten Vorschriften, Normen, Prozesse und Dokumentationen für die Gebrauchstauglichkeit von Medizinprodukten anzuwenden. Sie erfahren, wie Sie Anwender und Anwendungssituationen von Medizinprodukten erfolgreich in Ihren Entwicklungsprozess einbeziehen.
Unsere Lehrgänge und Weiterbildungen im Bereich 'Prozess- Qualitäts- und Risikomanagement' helfen Ihnen, die Qualität und Leistung Ihrer Medizinprodukte über den gesamten Produktlebenszyklus zu sichern, indem Sie die Anforderungen an das Managementsystem, die Prozesse und die Dokumentation erfüllen und überwachen.
Expert Quality Management Medical Devices International (TÜV).
Gesamtlehrgang oder Kompaktlehrgang
Um die regulatorische Konformität von Medizinprodukten sicherzustellen, benötigen Unternehmen ein effizientes Managementsystem, strukturierte und geregelte Prozesse sowie ein wirksames System von Vorbeugungs- und Korrekturmaßnahmen. Dazu gehören die Anwendung internationaler Managementnormen wie ISO 13485, das Risikomanagement nach ISO/EN ISO 14971:2019/EN ISO 14971:2019+A11:2021 und die Gebrauchstauglichkeit nach EN 62366, die Umsetzung der MDR und anderer Marktregeln sowie die Planung und Durchführung von Audits.
1st and 2nd Party Auditor Medical Devices International (TÜV).
Gesamtlehrgang oder Kompaktlehrgang
Dieser Lehrgang zeigt Ihnen, wie Sie Audits für Medizinprodukte durchführen, um die Qualität, Leistung und regulatorische Konformität über den Lebenszyklus zu sichern, auch bei ausgelagerten Prozessen und Lieferanten. Sie bauen mit diesem Lehrgang auf Ihrem Wissen als 'Expert Quality Management Medical Devices International (TÜV)' auf und lernen praxisorientiert die Auditvorbereitung, -durchführung und -nachbereitung – auch bei Remoteaudits wissen Sie, was Sie für die Online-Durchführung berücksichtigen müssen. Ideal auch für Quereinstieger die den EQM-Abschluss bereits besitzen und Auditor werden möchten!
Riskmanager Medical Devices International (TÜV).
Gesamtlehrgang
Medizinprodukte müssen während ihres gesamten Produktlebenszyklus von der Entwicklung bis zur Marktbeobachtung (Post Market Surveillance) ein systematisches Risikomanagement auf Basis der ISO 14971:2019 / EN ISO 14971:2019 durchlaufen. Unser modularer Lehrgang macht Sie mit den regulatorischen und normativen Rahmenbedingungen vertraut und zeigt Ihnen, wie Sie Ihr Risikomanagement praktisch umsetzen und dokumentieren können. In diesem Lehrgang vertiefen Sie ihr Wissen mit der Erarbeitung von Musterlösungen anhand von Praxisbeispielen.
Process Validation Expert Medical Devices (TÜV).
Gesamtlehrgang
Sowohl die QM-Norm ISO 13485 als auch die FDA-Spezifikation 21CFR820 fordern explizit die Validierung von Herstellungsprozessen, wenn die Prozessergebnisse im Rahmen der Herstellung von Medizinprodukten nicht verifizierbar sind. Mit der Prozessvalidierung lässt sich nachweisen, dass die Herstellprozesse stabil entwickelt wurden und zuverlässig arbeiten. In unserem Lehrgang werden Ihnen die relevanten Vorschriften, die gängigen Modelle und die statistischen Methoden für die Prozessvalidierung im Rahmen der Herstellung von Medizinprodukten vermittelt.
Renommiert: TÜV Rheinland steht für Qualität, Sicherheit und Neutralität. Der weltweit anerkannte Prüfdienstleister mit 20.000 Mitarbeiter in 65 Ländern bietet Ihnen branchenübergreifende Kompetenzen als Zertifizierer. Im Bereich Medizintechnik genießt die TÜV Rheinland Akademie seit vielen Jahren den Ruf als anerkannter Weiterbildungsanbieter, welcher neben den zahlreichen Seminaren auch regelmäßig Fachkonferenzen im Bereich Medizintechnik anbietet.
Transparenz: PersCert TÜV dokumentiert auf „Certipedia“, der Zertifikatsdatenbank von TÜV Rheinland, die Inhalte und den Umfang der aktuell bestehenden Zertifizierungsprogramme in der Personenzertifizierung und führt ein Register zertifizierter Personen. Durch die Darstellung der Prüfungsverfahren, ID‘s für jedes Zertifizierungsprogramm sowie QR-Codes, ist ein Höchstmaß an Nachvollziehbarkeit und Transparenz garantiert.
Unabhängigkeit: Die Trennung von Ausbildung und Prüfung bildet eine wesentliche Grundlage für die Unabhängigkeit unserer Zertifizierungsverfahren.
Vergleichbarkeit: Dokumentierte Verfahren sorgen dafür, dass Zertifizierungen weltweit auf vergleichbare Weise durchgeführt werden können. Dies ist die Basis für die konstante Qualität unserer Dienstleistung als Zertifizierer und ermöglicht den Vergleich bestätigter Kompetenzen auch über die Ländergrenzen hinaus. Es gilt insbesondere für Zertifizierungen, die im Geltungsbereich der DIN EN ISO 17024 erfolgen.
Nachhaltigkeit: Durch die Befristung der Zertifikate und Entwicklung von Zertifizierungsverfahren trägt Personenzertifizierung dazu bei, Kompetenzentwicklung nachhaltig aufzubauen. Wir bieten unseren Kunden diese Möglichkeiten und begleiten sie so kontinuierlich in ihrem weiteren beruflichen Werdegang.
Die Welt steht im ständigen Wandel. Ganze Berufsbilder ändern sich binnen kürzester Zeit. Änderungen und Fortschritte insbesondere in technischen Bereichen muss Rechnung getragen werden. Einem Zertifizierungsprogramm zugrundeliegende Standards müssen stets aktuell sein, Zertifizierungen auf Basis der aktuell geltenden Normen. Personen mit unbefristeter Zertifizierung, die längere Zeit nicht im zertifizierten Bereich tätig sind verlieren mit der Zeit ihre Handlungskompetenz und können zum (Sicherheits-)Risiko für den Arbeitgeber, andere oder sich selbst werden. Dies gilt es dringend zu vermeiden.
Die Rezertifzierung ist möglich, wenn innerhalb der drei Jahre mindestens ein Fortbildungstag absolviert und fortgesetzte abschlussbezogene Tätigkeit nachgewiesen wird. Durch die Teilnahme an unseren Weiterbildungen für die Medizintechnikbranche und unseren regelmäßig stattfindenden Medizinprodukte-Konferenzen erfüllen Sie die Voraussetzungen an die Fortbildung für die Rezertifizierung. Die genauen Informationen zur Rezertifizierung finden Sie in der jeweiligen Prüfungsordnung und auf unserer Rezert-Übersichtsseite. Antragsformulare sind unter www.certipedia.com herunter zu laden. Den Antrag zur Rezertifizierung sowie die erforderlichen Nachweise können Sie per E-Mail an den im Rezertifizierungsantrag benannten Ansprechpartner senden.
Ausführliche Informationen, welche Seminare sich für die Rezertifizierung Ihres PersCert TÜV-Abschlusses eignen, können Sie hier aufrufen.
Eine Rezertifizierung kann vom Zertifikatsinhaber spätestens bis zu 3 Monaten nach dem Ablauf der Gültigkeit des aktuellen Zertifikates schriftlich bei PersCert TÜV beantragt werden.
Die Zulassungsvoraussetzungen zur Teilnahme an den von PersCert TÜV, der unabhängigen Personenzertifizierungsstelle von TÜV Rheinland, durchgeführten Prüfungen, finden Sie auf www.certipedia.com. Spezifische Informationen und alle Starttermine finden Sie auch auf der jeweiligen Lehrgangs-Website.
Mit hochwertigen Webinaren, Whitepapers, Blogbeiträgen und Podcasts - zentral verfügbar im Medizintechnik-Wissensportal der TÜV Rheinland Akademie - erweitern Sie Ihr Medizintechnik-Know-how.
Ihr Feedback ist für uns von unschätzbarem Wert, denn es ermöglicht uns, unsere Kurse noch genauer auf Ihre Bedürfnisse und die Anforderungen des Marktes abzustimmen. Mit nur wenigen Klicks können Sie einen wichtigen Beitrag leisten und uns helfen, die Qualität und Relevanz unserer Bildungsangebote zu steigern.
Bitte nehmen Sie sich einen Moment Zeit um an unserer kurzen anonymen Umfrage teilzunehmen und damit die Chance zu nutzen, die Weiterbildungslandschaft in der Medizintechnik mitzugestalten.